CHAPTER 9
Chasing Memory
Understanding Alzheimer’s Disease and Other Neurodegenerative Diseases
The greatest obstacle to discovery is not ignorance—it is the illusion of knowledge.
—Daniel J. Boorstin
Most people tend to go to the doctor when they are sick or think they might be. Nearly all my patients first come to see me when they are relatively healthy, or think they are. That was the case with Stephanie, a forty-year-old woman who walked into my office for her initial visit in early 2018 with no real complaint. She was simply “interested” in longevity.
Her family history was not remarkable. Three of her four grandparents had died from complications of atherosclerosis in their late seventies or early eighties, and the fourth had died from cancer. Pretty much par for the course for the Greatest Generation. The only red flag was the fact that her mother, otherwise healthy at seventy, was beginning to suffer some memory loss, which Stephanie attributed to “old age.”
We set up another appointment for a week later to review her initial blood work. I rely as much as one can on biomarkers, so we run a comprehensive array of tests, but there are a few things that I immediately scan for when I get a new patient’s results back. Among them is their level of Lp(a), the high-risk lipoprotein that we talked about in chapter 7, along with their apoB concentration. A third thing that I always check is their APOE genotype, the gene related to Alzheimer’s disease risk that we mentioned in chapter 4.
Stephanie’s labs revealed that she had the APOE e4 allele, which is associated with a greater risk of Alzheimer’s disease—and not just one copy, but two (e4/e4), which meant her risk of developing Alzheimer’s disease was up to twelve times greater than that of someone with two copies of the common e3 allele. The e2 version of APOE appears to protect carriers against Alzheimer’s disease: 10 percent reduced risk for someone with e2/e3, and about 20 percent for e2/e2. Stephanie was unlucky.
She was only the fourth patient I’d ever encountered with this quite uncommon genotype, shared by only about 2 to 3 percent of the population, and she had no idea she was at risk—although in hindsight, her mother’s forgetfulness may have been a symptom of early Alzheimer’s disease. Now I faced a double challenge: how to break the news to her, directly but gently; and even more tricky, how to explain what it meant and what it didn’t mean.
In circumstances like this I generally think it’s best to get right to the point, so after we sat down, I said something like, “Stephanie, there is something we found in your blood test that may be of concern to me—not because anything is wrong now, but because of the risk it poses in twenty to thirty years or so. You have a combination of genes that increases your risk of developing Alzheimer’s disease. But it’s also important that you understand that what we’re about to discuss is only a marker for risk, not a fait accompli, and I am convinced that we can mitigate this risk going forward.”
Stephanie was devastated. She was dealing with a lot of stress to begin with: a divorce, a difficult work situation, and now this. It’s tough to explain the nuances of genes and risk to somebody when their eyes are wide with fear and they are hearing only I’m doomed. It took several discussions over the course of many weeks before she began to understand the rest of the message, which was that she was not, in fact, doomed.
Alzheimer’s disease is perhaps the most difficult, most intractable of the Horsemen diseases. We have a much more limited understanding of how and why it begins, and how to slow or prevent it, than we do with atherosclerosis. Unlike with cancer, we currently have no way to treat it once symptoms begin. And unlike type 2 diabetes and metabolic dysfunction, it does not appear to be readily reversible (although the jury is still out on that). This is why, almost without exception, my patients fear dementia more than any other consequence of aging, including death. They would rather die from cancer or heart disease than lose their minds, their very selves.
Alzheimer’s disease is the most common, but there are other neurodegenerative diseases that concern us. The most prevalent of these are Lewy body dementia and Parkinson’s disease, which are actually different forms of a related disorder known (confusingly) as “dementia with Lewy bodies.” The primary difference between them is that Lewy body dementia is primarily a dementing disorder, meaning it affects cognition, while Parkinson’s disease is considered primarily (but not entirely) a movement disorder, although it does also result in cognitive decline. In the United States, about 6 million people are diagnosed with Alzheimer’s disease, while about 1.4 million have Lewy body dementia, and 1 million have been diagnosed with Parkinson’s, which is the fastest-growing neurodegenerative disease. Beyond that, there are a variety of less common but also serious neurodegenerative conditions such as amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease) and Huntington’s disease.
All these result from some form of neurodegeneration, and as yet, there is no cure for any of them—despite the billions and billions of dollars that have been spent chasing these complex conditions. Maybe there will be a breakthrough in the near future, but for now our best and only strategy is to try to prevent them. The only shred of good news here is that while these disorders have traditionally been considered as completely separate and distinct diseases, evolving evidence suggests that there is a larger continuum between them than has previously been recognized, which means that some of our prevention strategies could apply to more than one of them as well.
Many doctors shy away from APOE gene testing. The conventional wisdom holds that someone with the high-risk e4 allele is all but guaranteed to develop Alzheimer’s disease, and there is nothing we can do for them. So why burden the patient with this terrible knowledge?
Because there are two types of bad news: that about things we can change, and about things that we can’t. Assuming that a patient’s e4 status falls into the latter category is, in my opinion, a mistake. While it is true that over one-half of people with Alzheimer’s disease have at least one copy of e4, merely possessing this risk gene is not the same as being diagnosed with dementia due to Alzheimer’s disease. There are e4/e4-carrying centenarians without any signs of dementia, likely because they have other genes that protect them from e4; for example, a certain variant of the gene Klotho (KL), called kl-vs, seems to protect carriers of e4 from developing dementia. And plenty of “normal” e3/e3 carriers will still go on to develop Alzheimer’s. Having the e4 gene variant merely signals increased risk. It’s not a done deal.
The other point I tried to make to Stephanie was that time was on her side. The disease rarely progresses to a clinical stage before about age sixty-five, even in patients with two copies of e4. That gives us about twenty-five years to try to prevent or delay her from developing this horrible illness with the tools currently at our disposal. In the interim, hopefully, researchers might come up with more effective treatments. It was a classic asymmetric situation, where doing nothing was actually the riskiest course of action.
Understanding Alzheimer’s
Although Alzheimer’s disease was first named in the early 1900s, the phenomenon of “senility” has been remarked on since ancient times. Plato believed that because advancing age seemingly “gives rise to all manners of forgetfulness as well as stupidity,” older men were unsuited for leadership positions requiring acumen or judgment. William Shakespeare gave us an unforgettable portrayal of an old man struggling with his failing mind in King Lear.
The notion that this might be a disease was first suggested by Dr. Alois Alzheimer, a psychiatrist who worked as medical director at the state asylum in Frankfurt, Germany. In 1906, while performing an autopsy on a patient named Auguste Deter, a woman in her midfifties who had suffered from memory loss, hallucinations, aggression, and confusion in her final years, he noticed that something was clearly wrong with her brain. Her neurons were entangled and spiderweb-like, coated with a strange white dental substance. He was so struck by their odd appearance that he made drawings of them.
Another colleague later dubbed this condition “Alzheimer’s disease,” but after Alzheimer himself died in 1915 (of complications from a cold, at fifty-one), the disease he had identified was more or less forgotten for fifty years, relegated to obscurity along with other less common neurological conditions such as Huntington’s and Parkinson’s diseases as well as Lewy body dementia. Patients with the kinds of symptoms that we now associate with these conditions, including mood changes, depression, memory loss, irritability, and irrationality, were routinely institutionalized, as Auguste Deter had been. Plain old “senility,” meanwhile, was considered to be an inevitable part of aging, as it had been since Plato’s day.
It wasn’t until the late 1960s that scientists began to accept that “senile dementia” was a disease state and not just a normal consequence of aging. Three British psychiatrists, Garry Blessed, Bernard Tomlinson, and Martin Roth, examined the brains of seventy patients who had died with dementia and found that many of them exhibited the same kinds of plaques and tangles that Alois Alzheimer had observed. Further studies revealed that a patient’s degree of cognitive impairment seemed to correlate with the extent of plaques found in his or her brain. These patients, they concluded, also had Alzheimer’s disease. A bit more than a decade later, in the early 1980s, other researchers identified the substance in the plaques as a peptide called amyloid-beta. Because it is often found at the scene of the crime, amyloid-beta was immediately suspected to be a primary cause of Alzheimer’s disease.
Amyloid-beta is a by-product that is created when a normally occurring substance called amyloid precursor protein, or APP, a membrane protein that is found in neuronal synapses, is cleaved into three pieces. Normally, APP is split into two pieces, and everything is fine. But when APP is cut in thirds, one of the resulting fragments then becomes “misfolded,” meaning it loses its normal structure (and thus its function) and becomes chemically stickier, prone to aggregating in clumps. This is amyloid-beta, and it is clearly bad stuff. Laboratory mice that have been genetically engineered to accumulate amyloid-beta (they don’t naturally) have difficulty performing cognitive tasks that are normally easy, such as finding food in a simple maze. At the same time, amyloid also triggers the aggregation of another protein called tau, which in turn leads to neuronal inflammation and, ultimately, brain shrinkage. Tau was likely responsible for the neuronal “tangles” that Alois Alzheimer observed in Auguste Deter.
Scientists have identified a handful of genetic mutations that promote very rapid amyloid-beta accumulation, all but ensuring that someone will develop the disease, often at a fairly young age. These mutations, the most common of which are called APP, PSEN1, and PSEN2, typically affect the APP cleavage. In families carrying these genes, very-early-onset Alzheimer’s disease is rampant, with family members often developing symptoms in their thirties and forties. Luckily, these mutations are very rare, but they occur in 10 percent of early-onset Alzheimer’s cases (or about 1 percent of total cases). People with Down syndrome also tend to accumulate large amounts of amyloid plaques over time, because of genes related to APP cleavage that reside on chromosome 21.
It was not a huge leap to conclude, on the basis of available evidence, that Alzheimer’s disease is caused directly by this accumulation of amyloid-beta in the brain. The “amyloid hypothesis,” as it’s called, has been the dominant theory of Alzheimer’s disease since the 1980s, and it has driven the research priorities of the National Institutes of Health and the pharmaceutical industry alike. If you could eliminate the amyloid, the thinking has been, then you could halt or even reverse the progression of the disease. But it hasn’t worked out that way. Several dozen drugs have been developed that target amyloid-beta in one way or another. But even when they succeed in clearing amyloid or slowing its production, these drugs have yet to show benefit in improving patients’ cognitive function or slowing the progression of the disease. Every single one of them failed.
One hypothesis that emerged, as these drugs were failing one after another, was that patients were being given the drugs too late, when the disease had already taken hold. It is well known that Alzheimer’s develops slowly, over decades. What if we gave the drugs earlier? This promising hypothesis has been tested in large and well-publicized clinical trials involving people with an inherited mutation that basically predestines them to early-onset Alzheimer’s, but those, too, failed in the end. A broader clinical trial was launched in 2022 by Roche and Genentech, testing early administration of an anti-amyloid compound in genetically normal people with verified amyloid accumulation in their brains but no clear symptoms of dementia; results are expected in 2026. Some researchers think that the disease process might be reversible at the point where amyloid is present but not tau, which appears later. That theory is being tested in yet another ongoing study.
Meanwhile, in June of 2021, the FDA gave approval to an amyloid-targeting drug called aducanumab (Aduhelm). The drug’s maker, Biogen, had submitted data for approval twice before and had been turned down. This was its third try. The agency’s expert advisory panel recommended against approving the drug this time as well, saying the evidence of a benefit was weak or conflicted, but the agency went ahead and approved it anyway. It has met a tepid reception in the marketplace, with Medicare and some insurers refusing to pay its $28,000 annual cost unless it is being used in a clinical trial at a university.
This cascade of drug failures has caused frustration and confusion in the Alzheimer’s field, because amyloid has long been considered a signature of the disease. As Dr. Ronald Petersen, director of the Mayo Clinic Alzheimer’s Disease Research Center, put it to The New York Times in 2020: “Amyloid and tau define the disease….To not attack amyloid doesn’t make sense.”
But some scientists have begun to openly question the notion that amyloid causes all cases of Alzheimer’s disease, citing these drug failures above all. Their doubts seemed to be validated in July of 2022, when Science published an article calling into question a widely cited 2006 study that had given new impetus to the amyloid theory, at a time when it had already seemed to be weakening. The 2006 study had pinpointed a particular subtype of amyloid that it claimed directly caused neurodegeneration. That in turn inspired numerous investigations into that subtype. But according to the Science article, key images in that study had been falsified.
There was already plenty of other evidence calling into question the causal relationship that has long been assumed between amyloid and neurodegeneration. Autopsy studies have found that more than 25 percent of cognitively normal people nevertheless had large deposits of amyloid in their brains when they died—some of them with the same degree of plaque buildup as patients who died with severe dementia. But for some reason, these people displayed no cognitive symptoms. This was not actually a new observation: Blessed, Tomlinson, and Roth noted back in 1968 that other researchers had observed “plaque formation and other changes [that] were sometimes just as intense in normal subjects as in cases of senile dementia.”
Some experts maintain that these patients actually did have the disease but that its symptoms had been slower to emerge, or that they had somehow masked or compensated for the damage to their brains. But more recent studies have found that the reverse can also be true: some patients with all the symptoms of Alzheimer’s disease, including significant cognitive decline, have little to no amyloid in their brains, according to amyloid PET scans and/or cerebrospinal fluid (CSF) biomarker testing, two common diagnostic techniques. Researchers from the Memory and Aging Center at University of California San Francisco found via PET scans that close to one in three patients with mild to moderate dementia had no evidence of amyloid in their brains. Still other studies have found only a weak correlation between the degree of amyloid burden and the severity of disease. It appears, then, that the presence of amyloid-beta plaques may be neither necessary for the development of Alzheimer’s disease nor sufficient to cause it.
This raises another possibility: that the condition that Alois Alzheimer observed in 1906 was not the same condition as the Alzheimer’s disease that afflicts millions of people around the world. One major clue has to do with the age of onset. Typically, what we call Alzheimer’s disease (or late-onset Alzheimer’s disease) does not present in significant numbers until age sixty-five. But Dr. Alzheimer’s own Patient Zero, Auguste Deter, showed severe symptoms by the time she was fifty, a trajectory more in line with early-onset Alzheimer’s disease than with the dementia that slowly begins to afflict people in their late sixties, seventies, and eighties. A 2013 analysis of preserved tissue from Auguste Deter’s brain found that she did in fact carry the PSEN1 mutation, one of the early-onset dementia genes. (It affects the cleavage of the amyloid precursor protein, producing loads of amyloid.) She did have Alzheimer’s disease, but a form of it that you get only because you have one of these highly deterministic genes. Our mistake might have been to assume that the other 99 percent of Alzheimer’s disease cases progress the way hers did.
This is not all that uncommon in medicine, where the index case for a particular disease turns out to be the exception rather than the rule; extrapolating from this one case can lead to problems and misunderstanding down the road. At the same time, if Auguste Deter’s illness had appeared when she was seventy-five instead of fifty, then perhaps it might not have seemed remarkable at all.
Just as Alzheimer’s disease is defined (rightly or wrongly) by accumulations of amyloid and tau, Lewy body dementia and Parkinson’s disease are associated with the accumulation of a neurotoxic protein called alpha-synuclein, which builds up in aggregates known as Lewy bodies (first observed by a colleague of Alois Alzheimer’s named Friedrich Lewy). The APOE e4 variant not only increases someone’s risk for Alzheimer’s but also significantly raises their risk of Lewy body dementia as well as Parkinson’s disease with dementia, further supporting the notion that these conditions are related on some level.
All this places high-risk patients like Stephanie in a terrible predicament: they are at increased risk of developing a disorder or disorders whose causes we still do not fully understand, and for which we lack effective treatments. That means we need to focus on what until fairly recently was considered a taboo topic in neurodegenerative disease: prevention.
Can Neurodegenerative Disease Be Prevented?
Stephanie was terrified. I had treated other patients who had two copies of the e4 gene, but none of them had responded with this much fear and anxiety. It took us four long discussions over a period of two months just to get her past the initial shock of the news. Then it was time to have a talk about what to do.
She had no obvious signs of cognitive impairment or memory loss. Yet. Just as a side note, some of my patients freak out because they lose their car keys or their phones from time to time. As I keep reminding them, that does not mean they have Alzheimer’s disease. It generally only means that they are busy and distracted (ironically, often by the very same cell phones that they keep misplacing). Stephanie was different. Her risk was real, and now she knew it.
With my highest-risk patients, like Stephanie, I would at that time typically collaborate with Dr. Richard Isaacson, who opened the first Alzheimer’s disease prevention clinic in the United States in 2013. Richard remembers that when he first interviewed with the dean of Weill Cornell Medical College and described his proposed venture, she seemed taken aback by his then-radical idea; the disease was not considered to be preventable. Also, at barely thirty, he did not look the part. “She was expecting Oliver Sacks,” he told me. “Instead, she was sitting across the table from Doogie Howser.”
Isaacson had gone off to college at age seventeen, to a joint BA-MD program at the University of Missouri in Kansas City and had his medical degree in hand by the time he was twenty-three. He was motivated to learn as much as possible about Alzheimer’s disease by the fear that it ran in his family.
When he was growing up in Commack, Long Island, Richard had watched his favorite relative, his great-uncle Bob, succumb to Alzheimer’s disease. When Richard was three, Uncle Bob had saved him from drowning in a swimming pool at a family party. But about a decade later, his beloved uncle had begun to change. He repeated his stories. His sense of humor faded. He developed a vacant stare and seemed to take longer to process what he was hearing around him. He was eventually diagnosed with Alzheimer’s disease—but he was already gone. It was as if he had vanished.
Richard couldn’t stand to watch his favorite uncle be reduced to nothing. He was also worried because he knew that Alzheimer’s disease risk has a strong genetic component. Was he also at risk? Were his parents? Could the disease somehow be prevented?
As he went into medical practice at the University of Miami, he began collecting every single study and recommendation he could find on possible ways to reduce risk of Alzheimer’s disease, gathering them into a photocopied sheaf that he would hand out to patients. He published the sheaf in 2010 as a book titled Alzheimer’s Treatment, Alzheimer’s Prevention: A Patient and Family Guide.
He soon found out that in Alzheimer’s disease circles, the very word prevention was somewhat off limits. “The Alzheimer’s Association said, you can’t really say this,” he remembers. That same year, a panel of experts assembled by the National Institutes of Health declared, “Currently, firm conclusions cannot be drawn about the association of any modifiable risk factor with cognitive decline or Alzheimer’s disease.”
That led Isaacson to realize that the only way to show that a preventive approach could work, in a way that would be accepted by the broader medical community, was in a large academic setting. Cornell was willing to take a chance, and his clinic opened in 2013. It was the first of its kind in the country, but now there are half a dozen similar centers, including one in Puerto Rico. (Isaacson now works for a private company in New York; his colleague, Dr. Kellyann Niotis, has joined my practice, focusing on patients at risk for neurodegenerative diseases.)
Meanwhile, the idea of preventing Alzheimer’s disease began to gain scientific support. A two-year randomized controlled trial in Finland, published in 2015, found that interventions around nutrition, physical activity, and cognitive training helped maintain cognitive function and prevent cognitive decline among a group of more than 1,200 at-risk older adults. Two other large European trials have found that multidomain lifestyle-based interventions have improved cognitive performance among at-risk adults. So there were signs of hope.
To a typical doctor, Stephanie’s case would have seemed pointless: She had no symptoms and she was still relatively young, in her early forties, a good two decades before any clinical dementia was likely to develop. Medicine 2.0 would say there was nothing for us to treat yet. In Medicine 3.0, this makes her an ideal patient, and her case an urgent one. If there were ever a disease that called for a Medicine 3.0 approach—where prevention is not only important but our only option—Alzheimer’s disease and related neurodegenerative diseases are it.
Frankly, it might seem odd for a practice as small as ours to employ a full-time preventive neurologist such as Kellyann Niotis. Why do we do this? Because we think we can really move the needle by starting early and being very rigorous in how we quantify and then try to address each patient’s risk. Some patients, like Stephanie, are at obvious higher risk; but in a broader sense, all of us are at some risk of Alzheimer’s disease and other neurodegenerative disease.
Viewed through the lens of prevention, the fact that Stephanie had the APOE e4/e4 genotype was actually good news, in a way. Yes, she was at far higher risk than someone with e3/e3, but at least we knew the genes we were up against and the likely trajectory of the disease, if she developed it. It is more worrying when a patient presents with an overwhelming family history of dementia, or the early signs of cognitive decline, but does not carry any of the known Alzheimer’s risk genes, such as APOE e4 and a few others. That means that there could be some other risk genes in play, and we don’t know what they might be. Stephanie was at least facing a known risk. This was a start.
She had two additional risk factors that were out of her control: being Caucasian and being female. While those of African descent are at an overall increased risk of developing Alzheimer’s disease, for unclear reasons, APOE e4 seems to present less risk to them than to people of Caucasian, Asian, and Hispanic descent. Regardless of APOE genotype, however, Alzheimer’s disease is almost twice as common in women than in men. It is tempting to attribute this to the fact that more women live to age eighty-five and above, where incidence of the disease pushes 40 percent. But that alone does not explain the differential. Some scientists believe there may be something about menopause, and the abrupt decline in hormonal signaling, that sharply increases the risk of neurodegeneration in older women. In particular, it appears that a rapid drop in estradiol in women with an e4 allele is a driver of risk; that, in turn, suggests a possible role for perimenopausal hormone replacement therapy in these women.
Menopause is not the only issue here. Other reproductive history factors, such as the number of children the woman has had, age of first menstruation, and exposure to oral contraceptives, may also have a significant impact on Alzheimer’s risk and later life cognition. And new research suggests that women are more prone to accumulate tau, the neurotoxic protein we mentioned earlier. The end result is that women have a greater age-adjusted risk of Alzheimer’s, as well as faster rates of disease progression overall, regardless of age and educational level.
While female Alzheimer’s patients outnumber men by two to one, the reverse holds true for Lewy body dementia and Parkinson’s, both of which are twice as prevalent in men. Yet Parkinson’s also appears to progress more rapidly in women than in men, for reasons that are not clear.
Parkinson’s is also tricky genetically: While we have identified several gene variants that increase risk for PD, such as LRRK2 and SNCA, about 15 percent of patients diagnosed have some family history of the disease—and are therefore presumed to have a genetic component—but do not have any known risk genes or SNPs.
Like the other Horsemen, dementia has an extremely long prologue. Its beginnings are so subtle that very often the disease isn’t recognized until someone is well into its early stages. This is when their symptoms go beyond occasional lapses and forgetfulness to noticeable memory problems such as forgetting common words and frequently losing important objects (forgetting passwords becomes a problem too). Friends and loved ones notice changes, and performance on cognitive tests begins to slip.
Medicine 2.0 has begun to recognize this early clinical stage of Alzheimer’s, which is known as mild cognitive impairment (MCI). But MCI is not the first stage on the long road to dementia: a large 2011 analysis of data from the UK’s Whitehall II cohort study found that subtler signs of cognitive changes often become apparent well before patients meet the criteria for MCI. This is called stage I preclinical Alzheimer’s disease, and in the United States alone over forty-six million people are estimated to be in this stage, where the disease is slowly laying the pathological scaffolding in and around neurons but major symptoms are still largely absent. While it’s not clear how many of these patients will go on to develop Alzheimer’s, what is clear is that just as most of an iceberg lies beneath the surface of the ocean, dementia can progress unnoticed for years before any symptoms appear.
The same is true of other neurodegenerative diseases, although they each have different early warning signs. Parkinson’s may show up as subtle changes in movement patterns, a frozen facial expression, stooped posture or shuffling gait, a mild tremor, or even changes in a person’s handwriting (which may become small and cramped). Someone in the early stages of Lewy body dementia may exhibit similar physical symptoms, but with slight cognitive changes as well; both may exhibit alterations in mood, such as depression or anxiety. Something seems “off,” but it’s hard for a layperson to pinpoint.
This is why an important first step with any patient who may have cognitive issues is to subject them to a grueling battery of tests. One reason I like to have a preventive neurologist on staff is that these tests are so complicated and difficult to administer that I feel they are best left to specialists. They are also critically important to a correct diagnosis—assessing whether the patient is already on the road to Alzheimer’s disease or to another form of neurodegenerative dementia, and how far along they might be. These are clinically validated, highly complex tests that cover every domain of cognition and memory, including executive function, attention, processing speed, verbal fluency and memory (recalling a list of words), logical memory (recalling a phrase in the middle of a paragraph), associative memory (linking a name to a face), spatial memory (location of items in a room), and semantic memory (how many animals you can name in a minute, for example). My patients almost always come back complaining about the difficulty of the tests. I just smile and nod.
The intricacies and nuances of the tests give us important clues about what might be happening inside the brains of patients who are still very early in the process of cognitive change that goes along with age. Most importantly, they enable us to distinguish between normal brain aging and changes that may lead to dementia. One important section of the cognitive testing evaluates the patient’s sense of smell. Can they correctly identify scents such as coffee, for example? Olfactory neurons are among the first to be affected by Alzheimer’s disease.
Specialists such as Richard and Kellyann also become attuned to other, less quantifiable changes in people on the road to Alzheimer’s disease, including changes in gait, facial expressions during conversations, even visual tracking. These changes could be subtle and not recognizable to the average person, but someone more skilled can spot them.
The trickiest part of the testing is interpreting the results to distinguish among different types of neurodegenerative disease and dementia. Kellyann dissects the test results to try and trace the likely location of the pathology in the brain, and the specific neurotransmitters that are involved; these determine the pathological features of the disease. Frontal and vascular dementias primarily affect the frontal lobe, a region of the brain responsible for executive functioning such as attention, organization, processing speed, and problem solving. So these forms of dementia rob an individual of such higher-order cognitive features. Alzheimer’s disease, on the other hand, predominantly affects the temporal lobes, so the most distinct symptoms relate to memory, language, and auditory processing (forming and comprehending speech)—although researchers are beginning to identify different possible subtypes of Alzheimer’s disease, based on which brain regions are most affected. Parkinson’s is a bit different in that it manifests primarily as a movement disorder, resulting from (in part) a deficiency in producing dopamine, a key neurotransmitter. While Alzheimer’s can be confirmed by testing for amyloid in the cerebrospinal fluid, these other forms of neurodegeneration are largely clinical diagnoses, based on testing and interpretation. Thus, they can be more subjective, but with all these conditions it is critical to identify them as soon as possible, to allow more time for preventive strategies to work.
One reason why Alzheimer’s and related dementias can be so tricky to diagnose is that our highly complex brains are adept at compensating for damage, in a way that conceals these early stages of neurodegeneration. When we have a thought or a perception, it’s not just one neural network that is responsible for that insight, or that decision, but many individual networks working simultaneously on the same problem, according to Francisco Gonzalez-Lima, a behavioral neuroscientist at the University of Texas in Austin. These parallel networks can reach different conclusions, so when we use the expression “I am of two minds about something,” that is not scientifically inaccurate. The brain then picks the most common response. There is redundancy built into the system.
The more of these networks and subnetworks that we have built up over our lifetime, via education or experience, or by developing complex skills such as speaking a foreign language or playing a musical instrument, the more resistant to cognitive decline we will tend to be. The brain can continue functioning more or less normally, even as some of these networks begin to fail. This is called “cognitive reserve,” and it has been shown to help some patients to resist the symptoms of Alzheimer’s disease. It seems to take a longer time for the disease to affect their ability to function. “People that have Alzheimer’s disease and are very cognitively engaged, and have a good backup pathway, they’re not going to decline as quickly,” Richard says.
There is a parallel concept known as “movement reserve” that becomes relevant with Parkinson’s disease. People with better movement patterns, and a longer history of moving their bodies, such as trained or frequent athletes, tend to resist or slow the progression of the disease as compared to sedentary people. This is also why movement and exercise, not merely aerobic exercise but also more complex activities like boxing workouts, are a primary treatment/prevention strategy for Parkinson’s. Exercise is the only intervention shown to delay the progression of Parkinson’s.
But it’s difficult to disentangle cognitive reserve from other factors, such as socioeconomic status and education, which are in turn linked to better metabolic health and other factors (also known as “healthy user bias”). Thus, the evidence on whether cognitive reserve can be “trained” or used as a preventive strategy, such as by learning to play a musical instrument or other forms of “brain training,” is highly conflicted and not conclusive—although neither of these can hurt, so why not?
The evidence suggests that tasks or activities that present more varied challenges, requiring more nimble thinking and processing, are more productive at building and maintaining cognitive reserve. Simply doing a crossword puzzle every day, on the other hand, seems only to make people better at doing crossword puzzles. The same goes for movement reserve: dancing appears to be more effective than walking at delaying symptoms of Parkinson’s disease, possibly because it involves more complex movement.
This was one thing that Stephanie, a high-performing, well-educated professional, had in her favor. Her cognitive reserve was very robust, and her baseline scores were strong. This meant that we likely had plenty of time to devise a prevention strategy for her, perhaps decades—but given her increased genetic risk, we could not afford to delay. We needed to come up with a plan. What would that plan look like? How can this seemingly unstoppable disease be prevented?
We’ll start by taking a closer look at changes that might be happening inside the brain of someone on the road to Alzheimer’s. How are these changes contributing to the progression of the disease, and can we do anything to stop them or limit the damage?
Once we begin looking at Alzheimer’s disease outside the prism of the amyloid theory, we start to see certain other defining characteristics of dementia that might offer opportunities for prevention—weaknesses in our opponent’s armor.
Alternatives to Amyloid
For decades, almost in tandem with observations of plaques and tangles, researchers have also noted problems with cerebral blood flow, or “perfusion,” in patients with dementia. On autopsy, Alzheimer’s brains often display marked calcification[*] of the blood vessels and capillaries that feed them. This is not a new observation: In their seminal 1968 paper that defined Alzheimer’s disease as a common age-related condition, Blessed, Tomlinson, and Roth had also noted severe vascular damage in the brains of their deceased study subjects. The phenomenon had been noted in passing for decades, as far back as 1927. But it was generally considered to be a consequence of neurodegeneration and not a potential cause.
In the early 1990s, a Case Western Reserve neurologist named Jack de la Torre was flying to Paris for a conference and thinking about the origins of Alzheimer’s disease. The amyloid hypothesis was still fairly new, but it didn’t sit well with de la Torre because of what he had observed in his own lab. On the flight, he had a eureka moment. “The evidence from dozens of rat experiments seemed to be screaming at me,” he later wrote. In those experiments, he had restricted the amount of blood flowing to the rats’ brains, and over time they had developed symptoms remarkably similar to those of Alzheimer’s disease in humans: memory loss and severe atrophy of the cortex and hippocampus. Restoring blood flow could halt or reverse the damage to some extent, but it seemed to be more severe and more lasting in older animals than younger ones. The key insight was that robust blood flow seemed to be critical to maintaining brain health.
The brain is a greedy organ. It makes up just 2 percent of our body weight, yet it accounts for about 20 percent of our total energy expenditure. Its eighty-six billion neurons each have between one thousand and ten thousand synapses connecting them to other neurons or target cells, creating our thoughts, our personalities, our memories, and the reasoning behind both our good and bad decisions. There are computers that are bigger and faster, but no machine yet made by man can match the brain’s ability to intuit and learn, much less feel or create. No computer possesses anything approaching the multidimensionality of the human self. Where a computer is powered by electricity, the beautiful machine that is the human brain depends on a steady supply of glucose and oxygen, delivered via a huge and delicate network of blood vessels. Even slight disruptions to this vascular network can result in a crippling or even fatal stroke.
On top of this, brain cells metabolize glucose in a different way from the rest of the body; they do not depend on insulin, instead absorbing circulating glucose directly, via transporters that essentially open a gate in the cell membrane. This enables the brain to take top priority to fuel itself when blood glucose levels are low. If we lack new sources of glucose, the brain’s preferred fuel, the liver converts our fat into ketone bodies, as an alternative energy source that can sustain us for a very long time, depending on the extent of our fat stores. (Unlike muscle or liver, the brain itself does not store energy.) When our fat runs out, we will begin to consume our own muscle tissue, then our other organs, and even bone, all in order to keep the brain running at all costs. The brain is the last thing to shut off.
As his plane crossed the Atlantic, de la Torre scribbled down his ideas on the only available writing surface, which happened to be an air-sickness bag. The flight attendants looked grim as he asked for another bag, and another. His “barf bag theory,” as he jokingly called it, was that Alzheimer’s disease is primarily a vascular disorder of the brain. The dementia symptoms that we see result from a gradual reduction in blood flow, which eventually creates what he calls a “neuronal energy crisis,” which in turn triggers a cascade of unfortunate events that harms the neurons and ultimately causes neurodegeneration. The amyloid plaques and tangles come later, as a consequence rather than a cause. “We believed, and still do, that amyloid-beta is an important pathological product of neurodegeneration,” de la Torre wrote recently, “…[but] it is not the cause of Alzheimer’s disease.”
There was already evidence to support his theory. Alzheimer’s is more likely to be diagnosed in patients who have suffered a stroke, which typically results from a sudden blockage of blood flow in specific regions of the brain. In these cases, symptoms emerge abruptly, as if a switch has been flipped. Additionally, it has been established that people with a history of cardiovascular disease are at a higher risk of developing Alzheimer’s disease. Evidence also demonstrates a linear relationship between cognitive decline and increased intimal media thickness in the carotid artery, a major blood vessel that feeds the brain. Cerebral blood flow already declines naturally during the aging process, and this arterial thickening, a measure of arterial aging, could cause a further reduction in cerebral blood supply. Vascular disease is not the only culprit here either. In all, some two dozen known risk factors for Alzheimer’s disease also happen to reduce blood flow, including high blood pressure, smoking, head injury, and depression, among others. The circumstantial evidence is strong.
Improved neuroimaging techniques have confirmed not only that cerebral perfusion is decreased in brains affected by Alzheimer’s disease but also that a drop in blood flow seems to predict when a person will transition from preclinical Alzheimer’s disease to MCI, and on to full-fledged dementia. Although vascular dementia is currently considered distinct from dementia due to Alzheimer’s, making up roughly 15 to 20 percent of dementia diagnoses in North America and Europe, and up to 30 percent in Asia and developing countries, its symptoms and pathology overlap so significantly that de la Torre considers them different manifestations of the same basic condition.
Another compelling and perhaps parallel theory of Alzheimer’s disease says that it stems from abnormal glucose metabolism in the brain. Scientists and physicians have long noted a connection between Alzheimer’s disease and metabolic dysfunction. Having type 2 diabetes doubles or triples your risk of developing Alzheimer’s disease, about the same as having one copy of the APOE e4 gene. On a purely mechanistic level, chronically elevated blood glucose, as seen in type 2 diabetes and prediabetes/insulin resistance, can directly damage the vasculature of the brain. But insulin resistance alone is enough to elevate one’s risk
Insulin seems to play a key role in memory function. Insulin receptors are highly concentrated in the hippocampus, the memory center of the brain. Several studies have found that spraying insulin right into subjects’ noses—administering it as directly as possible into their brains—quickly improves cognitive performance and memory, even in people who have already been diagnosed with Alzheimer’s disease. One study found that intranasal insulin helped preserve brain volume in Alzheimer’s patients. Clearly, it is helpful to get glucose into neurons; insulin resistance blocks this. As the authors wrote, “Several lines of evidence converge to suggest that central insulin resistance plays a causal role in the development and progression of Alzheimer’s disease.”
The signal event here (again) appears to be a drop in energy delivery to the brain, similar to what is seen in the onset of vascular dementia. Brain imaging studies reveal lower brain glucose metabolism, decades before the onset of other symptoms of vascular dementia. Intriguingly, this reduction appears to be especially dramatic in brain regions that are also affected in Alzheimer’s disease, including the parietal lobe, which is important for processing and integrating sensory information; and the hippocampus of the temporal lobe, which is critical to memory. Just like reduced blood flow, reduced glucose metabolism essentially starves these neurons of energy, provoking a cascade of responses that include inflammation, increased oxidative stress, mitochondrial dysfunction—and ultimately neurodegeneration itself.
The Role of APOE e4
It is still not completely clear how or why, but e4 seems to accelerate other risk factors and driver mechanisms for Alzheimer’s—particularly metabolic factors such as reduced brain glucose metabolism, which we’ve just discussed. Simply put, it appears to make everything worse, including the Alzheimer’s gender gap: a woman with one copy of e4 is four times more likely to develop the disease than a man with the same genotype.
The protein for which it codes, APOE (apolipoprotein E), plays an important role in both cholesterol transport and glucose metabolism. It serves as the main cholesterol carrier in the brain, moving cholesterol across the blood-brain barrier to supply the neurons with the large amounts of it they require. Hussain Yassine, a neuroscientist at the University of Southern California who studies the role of APOE in Alzheimer’s disease, compares its role to that of an orchestra conductor. For some reason, he says, people with the e4 allele appear to have defects in both cholesterol transport and glucose metabolism, to a degree not seen in those with e2 or e3. Even though the higher risk APOE e4 protein differs from the harmless e3 one by just one amino acid, it appears to be less efficient at moving cholesterol into and especially out of the brain. There is also some evidence that the APOE e4 protein may also cause early breakdown of the blood-brain barrier itself, making the brain more susceptible to injury and eventual degeneration.
Curiously, APOE e4 was not always a bad actor. For millions of years, all our post-primate ancestors were e4/e4. It was the original human allele. The e3 mutation showed up about 225,000 years ago, while e2 is a relative latecomer, arriving only in the last 10,000 years. Data from present-day populations with a high prevalence of e4 suggest that it may have been helpful for survival in environments with high levels of infectious disease: children carrying APOE e4 in Brazilian favelas are more resistant to diarrhea and have stronger cognitive development, for example. In environments where infectious disease was a leading cause of death, APOE e4 carriers may have been the lucky ones, in terms of longevity.
This survival benefit may have been due to the role of APOE e4 in promoting inflammation, which can be beneficial in some situations (e.g., fighting infection) but harmful in others (e.g., modern life). As we saw in chapter 7, inflammation promotes atherosclerotic damage to our blood vessels, setting the stage for Alzheimer’s disease and dementia. People with Alzheimer’s disease often have high levels of inflammatory cytokines such as TNF-alpha and IL-6 in their brains, and studies have also found higher levels of neuroinflammation in e4 carriers. None of these, obviously, are good for our long-term brain health; as noted earlier, e4 just seems to make every risk factor for Alzheimer’s disease worse.
The e4 variant also seems to be maladaptive in other ways, such as in dealing with our modern diets. Not only are e4 carriers more likely to develop metabolic syndrome in the first place, but the APOE e4 protein may be partially responsible for this, by disrupting the brain’s ability to regulate insulin levels and maintain glucose homeostasis in the body. This phenomenon becomes apparent when these patients are on continuous glucose monitoring, or CGM (which we’ll discuss in more detail in chapter 15). Even young patients with e4 show dramatic blood glucose spikes after eating carbohydrate-rich foods, although the clinical significance of this is unclear.
Thus, e4 itself could help drive the very same metabolic dysfunction that also increases risk of dementia. At the same time, it appears to intensify the damage done to the brain by metabolic dysfunction. Researchers have found that in high-glucose environments, the aberrant form of the APOE protein encoded by APOE e4 works to block insulin receptors in the brain, forming sticky clumps or aggregates that prevent neurons from taking in energy.
But not everyone with the APOE e4 genotype is affected by it in the same way. Its effects on disease risk and the course of disease are highly variable. Factors like biological sex, ethnicity, and lifestyle clearly play a role, but it is now believed that Alzheimer’s risk and the effect of APOE are also powerfully dependent on other Alzheimer’s-risk-related genes that a person might carry, such as Klotho, the protective gene we mentioned earlier. This could explain, for example, why some people with e4 may never go on to develop Alzheimer’s disease, while others do so quickly.
All this suggests that metabolic and vascular causes of dementia may be somewhat overlapping, just as patients with insulin resistance are also prone to vascular disease. And it tells us that with high-risk patients like Stephanie, we need to pay special attention to their metabolic health.
The Preventive Plan
In spite of everything, I remain cautiously optimistic for patients like Stephanie, even with her highly elevated genetic risk. The very concept of Alzheimer’s prevention is still relatively new; we have only begun to scratch the surface of what might be accomplished here. As we better understand the disease, our treatments and interventions can become more sophisticated and hopefully effective.
I actually think we know more about preventing Alzheimer’s than we do about preventing cancer. Our primary tool for preventing cancer is to not smoke and to keep our metabolic health on track, but that’s a very broad-brush approach that only takes us so far. We still need to screen aggressively and hope we somehow manage to find any cancers that do develop before it’s too late. With Alzheimer’s disease, we have a much larger preventive tool kit at our disposal, and much better diagnostic methods as well. It’s relatively easy to spot cognitive decline in its early stages, if we’re looking carefully. And we’re learning more about genetic factors as well, including those that at least partially offset high-risk genes like APOE e4.
Because metabolism plays such an outsize role with at-risk e4 patients like Stephanie, our first step is to address any metabolic issues they may have. Our goal is to improve glucose metabolism, inflammation, and oxidative stress. One possible recommendation for someone like her would be to switch to a Mediterranean-style diet, relying on more monounsaturated fats and fewer refined carbohydrates, in addition to regular consumption of fatty fish. There is some evidence that supplementation with the omega-3 fatty acid DHA, found in fish oil, may help maintain brain health, especially in e4/e4 carriers. Higher doses of DHA may be required because of e4-induced metabolic changes and dysfunction of the blood-brain barrier.
This is also one area where a ketogenic diet may offer a real functional advantage: when someone is in ketosis, their brain relies on a mix of ketones and glucose for fuel. Studies in Alzheimer’s patients find that while their brains become less able to utilize glucose, their ability to metabolize ketones does not decline. So it may make sense to try to diversify the brain’s fuel source from only glucose to both glucose and ketones. A systematic review of randomized controlled trials found that ketogenic therapies improved general cognition and memory in subjects with mild cognitive impairment and early-stage Alzheimer’s disease. Think of it as a flex-fuel strategy.
In Stephanie’s case, she cut out not only added sugar and highly refined carbohydrates but also alcohol. The precise role of alcohol in relation to Alzheimer’s disease remains somewhat controversial: some evidence suggests that alcohol may be slightly protective against Alzheimer’s, while other evidence shows that heavier drinking is itself a risk factor for the disease, and e4 carriers may be more susceptible to alcohol’s deleterious effects. I’m inclined to err on the side of caution, and so is Stephanie.
The single most powerful item in our preventive tool kit is exercise, which has a two-pronged impact on Alzheimer’s disease risk: it helps maintain glucose homeostasis, and it improves the health of our vasculature. So along with changing Stephanie’s diet, we put her back on a regular exercise program, focusing on steady endurance exercise to improve her mitochondrial efficiency. This had a side benefit in that it helped manage her off-the-charts high cortisol levels, due to stress; stress and anxiety-related risk seem more significant in females. As we’ll see in chapter 11, endurance exercise produces factors that directly target regions of the brain responsible for cognition and memory. It also helps lower inflammation and oxidative stress.
Strength training is likely just as important. A study looking at nearly half a million patients in the United Kingdom found that grip strength, an excellent proxy for overall strength, was strongly and inversely associated with the incidence of dementia (see figure 8). People in the lowest quartile of grip strength (i.e., the weakest) had a 72 percent higher incidence of dementia, compared to those in the top quartile. The authors found that this association held up even after adjusting for the usual confounders such as age, sex, socioeconomic status, diseases such as diabetes and cancer, smoking, and lifestyle factors such as sleep patterns, walking pace, and time spent watching TV. And there appeared to be no upper limit or “plateau” to this relationship; the greater someone’s grip strength, the lower their risk of dementia.
It’s tempting to dismiss findings like these for the same reasons we should be skeptical of epidemiology. But unlike epidemiology in nutrition (much more on that in chapter 14), the epidemiology linking strength and cardiorespiratory fitness to lower risk for neurodegeneration is so uniform in its direction and magnitude that my own skepticism of the power of exercise, circa 2012, has slowly melted away. I now tell patients that exercise is, full stop and hands down, the best tool we have in the neurodegeneration prevention tool kit. (We’ll explore the ins and outs of this in great detail in chapters 11 and 12.)
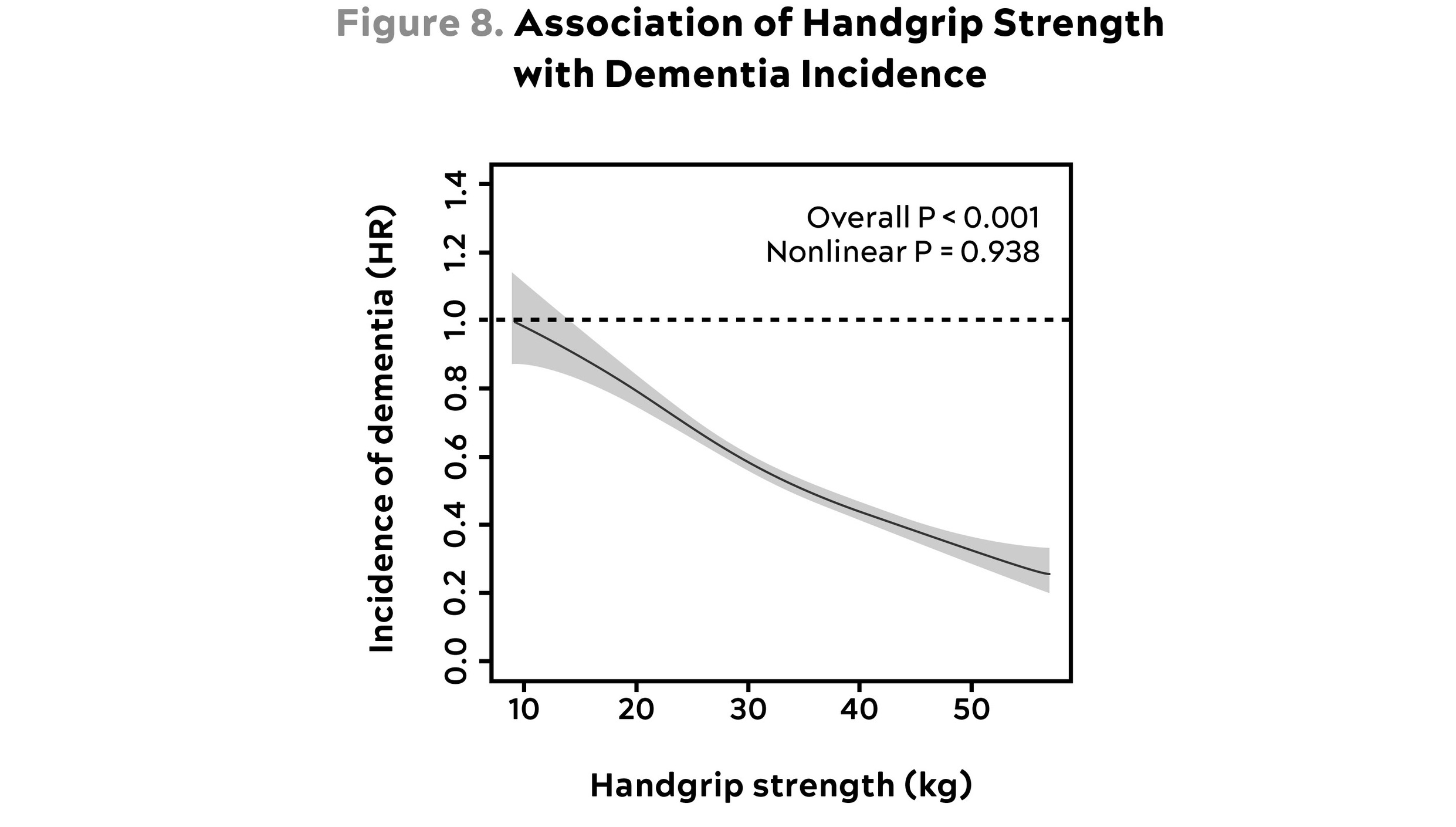
Source: Esteban-Cornejo et al. (2022).
This graph shows how the incidence of dementia declines with increasing handgrip strength. Note that data are presented as hazard ratios in comparison with the weakest group; e.g., 0.4 = 40 percent. Thus someone with 40 kg grip strength has about 40 percent as much risk of dementia as someone with 10 kg.
Sleep is also a very powerful tool against Alzheimer’s disease, as we’ll see in chapter 16. Sleep is when our brain heals itself; while we are in deep sleep our brains are essentially “cleaning house,” sweeping away intracellular waste that can build up between our neurons. Sleep disruptions and poor sleep are potential drivers of increased risk of dementia. If poor sleep is accompanied by high stress and elevated cortisol levels, as in Stephanie’s case, that acts almost as a multiplier of risk, as it contributes to insulin resistance and damaging the hippocampus at the same time. Furthermore, hypercortisolemia (excess cortisol due to stress) impairs the release of melatonin, the hormone that normally signals to our brains that it is time to go to sleep (and that may also help prevent neuronal loss and cognitive impairment). Addressing Stephanie’s difficulties with sleep was therefore urgent. Her divorce and her work situation were making it almost impossible for her to get more than four hours of uninterrupted sleep on any given night.
Another somewhat surprising risk factor that has emerged is hearing loss. Studies have found that hearing loss is clearly associated with Alzheimer’s disease, but it’s not a direct symptom. Rather, it seems hearing loss may be causally linked to cognitive decline, because folks with hearing loss tend to pull back and withdraw from interactions with others. When the brain is deprived of inputs—in this case auditory inputs—it withers. Patients with hearing loss miss out on socializing, intellectual stimulation, and feeling connected; prescribing them hearing aids may help relieve some symptoms. This is just a hypothesis for the moment, but it is being tested right now in a clinical trial called ACHIEVE (Aging and Cognitive Health Evaluation in Elders) that is currently ongoing.
While depression is also associated with Alzheimer’s disease, it appears to be more of a symptom than a risk factor or driver of the disease. Nevertheless, treating depression in patients with MCI or early Alzheimer’s disease does appear to help reduce some other symptoms of cognitive decline.
Another surprising intervention that may help reduce systemic inflammation, and possibly Alzheimer’s disease risk, is brushing and flossing one’s teeth. (You heard me: Floss.) There is a growing body of research linking oral health, particularly the state of one’s gum tissue, with overall health. Researchers have found that one pathogen in particular, a microbe called P. gingivalis that commonly causes gum disease, is responsible for large increases in levels of inflammatory markers such as IL-6. Even stranger, P. gingivalis has also shown up inside the brains of patients with Alzheimer’s disease, although scientists are not certain that this bacterium is directly causing dementia, notes Dr. Patricia Corby, a professor of dental health at New York University. Nevertheless, the association is too strong to be ignored. (Also, better oral health correlates strongly with better overall health, particularly in terms of cardiovascular disease risk, so I pay much more attention to flossing and gum health than I used to.)
One other somewhat recent addition to my thinking on dementia (and ASCVD while we’re at it) prevention is the use of dry saunas. Until about 2019 I was very skeptical of the data linking sauna use to brain and heart health. However, the more time I spend buried in this literature, the more I become convinced by the magnitude of the benefit, the uniformity of the studies, and the mechanisms providing plausibility. I’m not quite as confident that regular sauna use will reduce your risk of Alzheimer’s disease as I am that exercise will do so, but I am much more confident than I was at the outset of my journey. The best interpretation I can draw from the literature suggests that at least four sessions per week, of at least twenty minutes per session, at 179 degrees Fahrenheit (82 degrees Celsius) or hotter seems to be the sweet spot to reduce the risk of Alzheimer’s by about 65 percent (and the risk of ASCVD by 50 percent).
Other potential interventions that have shown some promise in studies include lowering homocysteine with B vitamins, while optimizing omega-3 fatty acids. Higher vitamin D levels have been correlated with better memory in e4/e4 patients but it’s difficult to know from the current literature if this means supplementing with vitamin D will reduce risk of AD. And as mentioned earlier, hormone replacement therapy for women during the transition from perimenopause to menopause seems promising, especially for women with at least one copy of e4.
The scariest aspect of Alzheimer’s disease boils down to this: Medicine 2.0 cannot help us. At all. The point at which Medicine 2.0 steps in, the point of diagnosis, is also likely near the point of no return for most Alzheimer’s patients, beyond which little or nothing can be done. Once dementia is diagnosed, it is extremely difficult to slow and maybe impossible to reverse (though we’re not certain of that). So we are forced to leave the familiar territory of the medicine that we know, with its promise of certainty, and embrace the Medicine 3.0 concepts of prevention and risk reduction.
As it stands now, Alzheimer’s disease is the last of the Horsemen that we must bypass on our way to becoming centenarians; it’s the last obstacle we face. Typically, it is diagnosed later in life—and centenarians develop it much later in life, if at all. The longer we can go without developing dementia, the better our odds of living longer, and living in better health. (Remember, cognition is one of the three key vectors of healthspan.) But until science comes up with more effective treatments, prevention is our only option. Therefore, we need to adopt a very early and comprehensive approach to preventing Alzheimer’s and other forms of neurodegenerative disease.
Broadly, our strategy should be based on the following principles:
-
WHAT’S GOOD FOR THE HEART IS GOOD FOR THE BRAIN. That is, vascular health (meaning low apoB, low inflammation, and low oxidative stress) is crucial to brain health.
-
WHAT’S GOOD FOR THE LIVER (AND PANCREAS) IS GOOD FOR THE BRAIN. Metabolic health is crucial to brain health.
-
TIME IS KEY. We need to think about prevention early, and the more the deck is stacked against you genetically, the harder you need to work and the sooner you need to start. As with cardiovascular disease, we need to play a very long game.
-
OUR MOST POWERFUL TOOL FOR PREVENTING COGNITIVE DECLINE IS EXERCISE. We’ve talked a lot about diet and metabolism, but exercise appears to act in multiple ways (vascular, metabolic) to preserve brain health; we’ll get into more detail in Part III, but exercise—lots of it—is a foundation of our Alzheimer’s-prevention program.
I have great hope that in the future we will learn much more about how to prevent and treat all forms of dementia. But it’s going to take hard work and creative thinking from scientists researching the disease, a significant investment in new theories and approaches, much more attention to strategies of prevention, and courage on the part of patients such as Stephanie who must face down this most feared and least understood of all the Horsemen.